Development of multiparent population resources

Our lab is part of a collaborative project to develop a community resource for the genetic dissection of complex traits in Drosophila melanogaster. The Drosophila Synthetic Population Resource (DSPR) is a multiparental population (MPP) consisting of a large collection of recombinant inbred lines derived from an 8-way 50 generation intercross. This design creates a panel of lines whose genomes are a mosaic of the original 8 founder lines, allowing for unprecedented mapping resolution for a linkage-based panel. In addition to using these lines in our research, we also work on describing the genetic properties of the resource and developing analytical tools and methodologies to enable other users. We have also developed and maintain the DSPRqtl R package to aid QTL mapping analyses with these lines.

Statistical Properties of the DSPR
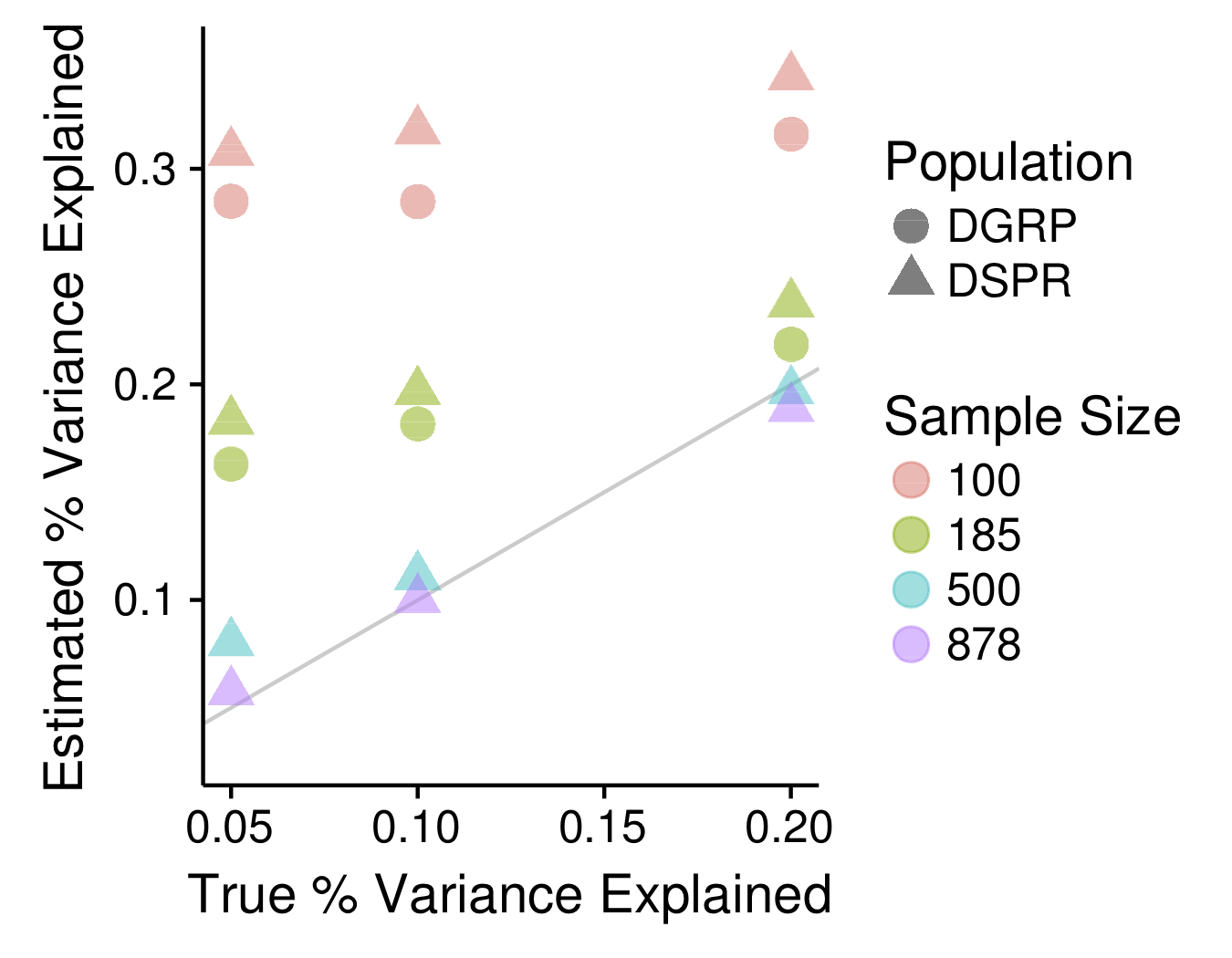
One of the goals in my lab is to investigate fundamental questions surrounding the statistical properties of MPPs generally, and the DSPR specifically. For example, in collaboration with Dr. Anthony Long's lab, we've used simulations to show how the Beavis effect, a statistical bias well-known in traditional QTL mapping approaches in which effect sizes for significant QTL tend to be overestimated, applies to modern mapping populations such as MPPs and genome-wide association (GWAS) panels. We've quantified the severity of this bias in both the DSPR and the DGRP, a GWAS-based D. melanogaster panel and used these results to provide guidelines for the experimental designs that will lead to the best estimates of effect size and the best prospects for validating significant QTL.
Inference of Haplotype Frequencies in Pooled Sequencing Approaches
One approach to the genetic dissection of complex phenotypes is a pooled sequencing approach, comparing pools of individuals that differ in some phenotype of interest and identifying regions of the genome that show differentiation between these pools. We have used this approach with an admixed population of DSPR RILs. In such a population, with known ancestry, it is possible to infer haplotype frequencies from sets of allele frequencies, which increases the power to identify differentiated regions. Our lab is developing optimization methods for inferring these haplotype frequencies. We've used the DSPR to simulate data to test the performance of these methods for different genomic window sizes, different sample sizes, and different sequence depth coverage to produce guidelines for the design of pooled sequencing experiments using MPPs.
